Protein Structural
Characterization
High Resolution Analysis of Protein & Antibody Structure
Hydrogen/deuterium exchange mass spectrometry (HDX-MS) has now become an indispensable technique for the structural and functional characterization of proteins.
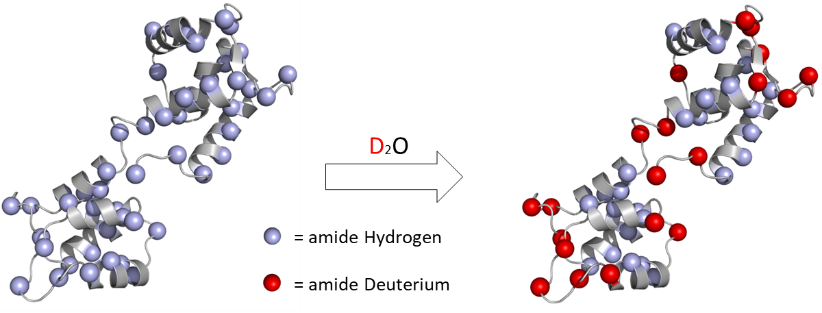
In a typical HDX workflow, a protein sample is immersed in D2O, at which time labile hydrogens (e.g., on the amides) begin to exchange with deuterium from the solvent. This exchange process is structure sensitive – the hydrogens on residues that are protected from solvent or involved in stable hydrogen-bonding exchange at a much slower rate. Specialized techniques in mass spectrometry can be used to reveal the detailed pattern of deuterium occupancy on individual residues. This information can be used to elucidate or confirm secondary structure, identify binding sites, reveal conformational changes and compare the structural similarity of antibodies or protein therapeutics.
Technological Advances
MRM Proteomics specializes in “top-down” HDX-MS. Existing HDX approaches commonly use “bottom-up” methods that require digestion of the protein prior to analysis. Our top-down approach bypasses this digestion step, so that the intact protein can be ionized and fragmented in the gas phase. By analyzing intact proteins instead of peptides, it is possible to achieve complete sequence coverage, residue-level resolution (instead of peptide-level resolution), and to minimize signal loss associated with back-exchange of deuterium atoms.
We have further optimized this HDX approach using subzero temperatures to minimize back exchange and specialized instrumentation to maximize coverage and generate comprehensive residue-by-residue information about protein structure. Our top-down HDX technologies are now compatible with proteins up to 150 KDa, including intact antibodies that were much too large to be analyzed by previous methods. This approach is extremely valuable for fine-grained characterization of biosimilars or antibodies. Our top-down methods can also be used for PTM analysis (including rapid global glycosylation profiling).

Features
Higher-order structural characterization of proteins including hydrogen bonding patterns
Detailed structural information with close-to-single-residue resolution and high sequence-coverage
Pinpoint conformational changes, bonding patterns, or binding sites in comparative studies
Generate isoform-specific information not available through bottom-up methods
Analyze intact antibodies up to 150 kDa using “top-down” or “middle-down” approaches
Additional structural and binding information can be generated through supplementary structural proteomics techniques (crosslinking, surface modification, etc.)
Pinpoint conformational changes, bonding patterns, or binding sites in comparative studies
Additional Tools & Techniques for Structural Proteomics
-
Features & Benefits
Higher-order structural characterization – reveals hydrogen bonding patterns and solvent exposure
Close-to-single-residue resolution
High sequence-coverage
Reduced back-exchange
Isoform-specific analysis
Applications
Analyze proteins & antibodies <150 kDa
Compare biosimilars to originator drugs
Pinpoint conformational changes, bonding patterns, or binding sites
-
Features & BenefitsHigher-order structural characterization
Reveals hydrogen bonding patterns
No size limitation on the proteins that can be analyzed
Applications
Analyze proteins & antibodies <150 kDa
Compare biosimilars to originator drugs
Pinpoint conformational changes, bonding patterns, or binding sites
-
Features & BenefitsCharacterize post-translational modifications (PTMs)
Generate isoform-specific information
Applications
Global glycosylation profiling
Determine disulphide bonding patterns
-
Features & BenefitsUltrahigh resolution
Applications
Determine mass of intact protein with extremely high accuracy
-
Features & BenefitsApproaching single-residue resolution
Applications
Determine or confirm primary structure
Applications
Clinical applications
Development of highly robust clinical tests to be used for improving diagnosis, risk stratification and treatment matching
Development of assays for monitoring treatment response/resistance, toxicity, disease progression
-
Features & BenefitsCharacterize 3D protein structures (tertiary & quaternary) for difficult-to-crystalize proteins
>10 custom reagents for surface modification
Applications
Analyze protein folding or misfolding
Determine protein complex assemblys
Generate additional constraint data for 3D structure modelings
-
Features & BenefitsCharacterize 3D protein structures (tertiary & quaternary)
Applications
Analyze protein folding or misfolding
Determine protein complex assemblys
Generate additional constraint data for 3D structure modelings
-
Features & BenefitsMultiple methods for lots of constraint data (crosslinking, surface medication, limited proteolysis, etc.)
Recent high profile publications for protein structure determination
Applications
Determine tertiary and quaternary structure for difficult-to-crystalize proteins
Map binding sites and orientations
Applications
Many new drugs are “biologics,” meaning that they are proteins or antibodies produced in biological expressions systems, rather than the more traditional small molecule drug entities produced by organic chemists and chemical synthesis. Part of the approval process for any drug, whether it is a biological or small molecule entity is for the manufacturer to prove that it has fully characterized the structure, purity, etc. of the drug compound. Generic drugs (called biosimilars in the case of biologics) have additional requirements: a generic or biosimilar manufacturer must prove that it has synthesized a drug molecule that has the same structure as the original (originator) drug.
This is very difficult for antibodies which have complex structures but are too small for direct imaging. Our clients have used the HDX-MS data generated by MRM Proteomics to confirm that the structure of a biosimilar protein drug matches the original biologic. These results have been used in FDA filings. HDX and other structural proteomics approaches have been successfully combined to fully elucidate the structure of high-molecular-weight protein complexes for which there was no high quality x-ray crystallography data (e.g., RNA polymerase II-Mediator core initiation complex). There are almost limitless possibilities for the application of structural proteomics techniques for investigating protein structure, function, and interactions.